
Genomisk selektion på gran

Inom förädlingen av både jordbruksgrödor och husdjur används idag rutinmässig tillämpning av genomisk selektion. Genomisk selektion innebär att man med hjälp av genetiska markörer, framför allt SNP (en punktmutation av ett enskilt baspar i DNA:t), tar fram de faktiska släktskapsrelationerna för de individer man arbetar med och sedan använder den informationen för urval i stället för använda både stamtavla och de mätta egenskaperna (fenotypmätningar).
Tyvärr har barrträden väldigt stor arvsmassa, vilket innebär att det varit både svårt och dyrt att utveckla metoden för tillämpningar inom skogsträdförädling. Utvecklingen har gått framåt de senaste åren, till exempel har stora mängder markördata tagits fram och granens arvsmassa har sekvenserats, samtidigt som kostnaden för sekvensering snabbt har minskat. Idag pågår ett flertal stora projekt inom vilka metoden utvecklas för många av våra trädslag. För att skogsträdsförädlingen fullt ut ska kunna använda sig av metoden så behöver även information från SNP. Detta skapar en variation mellan individer. kunna användas i de avancerade system för avelsutvärderingar som finns, exempelvis i ”Treeplan” som används inom skogsträdsförädlingen i Sverige.
Trädens egenskaper beräknas
Hur bra ett träd är i någon egenskap beror till stor del på vilken miljö det växer i, men även på vilka föräldrar det har, det vill säga vilka gener som har nedärvts. Inom skogsträdsförädlingen är fokuset på det genetiska bidraget för olika egenskaper som nedärvs från generation till generation. För att ta reda på vilka trädindivider som har de bästa förutsättningarna att generera avkommor som är bra i de egenskaper som är eftertraktade så anläggs fältförsök. Träden i försöken mäts efter ett antal år samtidigt som släktskapet mellan individerna utifrån stamtavlan används för att beräkna avelsvärden. Släktskapet beräknas traditionellt utifrån hur stor sannolikhet det är för två individer att dela DNA, till exempel så har helsyskon, som har samma mor och far, släktskapet 0,5. Ett annat sätt att beräkna släktskapet är med hjälp av genetiska markörer. I detta fall är det möjligt att även på små plantor beräkna släktskapet genom att studera hur stor del av genomet som är gemensamt mellan två individer. Den informationen kan sedan användas för att göra tidiga urval av de bästa individerna. Det tredje sättet att beräkna släktskapet är att kombinera de två tidigare nämnda sätten, det vill säga att för de individer där markördata finns tillgängliga används dessa, men i övrigt används det traditionella släktskapsförhållandet baserat på stamtavlan.
Avsikten med den här studien var att som ett första steg undersöka möjligheterna för skogsträdsförädlingen att kunna implementera genomisk selektion utvärderades precisionen för avelsvärden baserade på markördata. Detta gjordes med data från två försöksserier, en serie med fältförsök med trädindivider med ursprung från Västerbotten och en serie med ursprung från södra Norrland. Resultaten från en avelsvärdering där traditionella släktskapsförhållanden användes jämfördes med en avelsvärderingen där även genetiska markördata inkluderades.
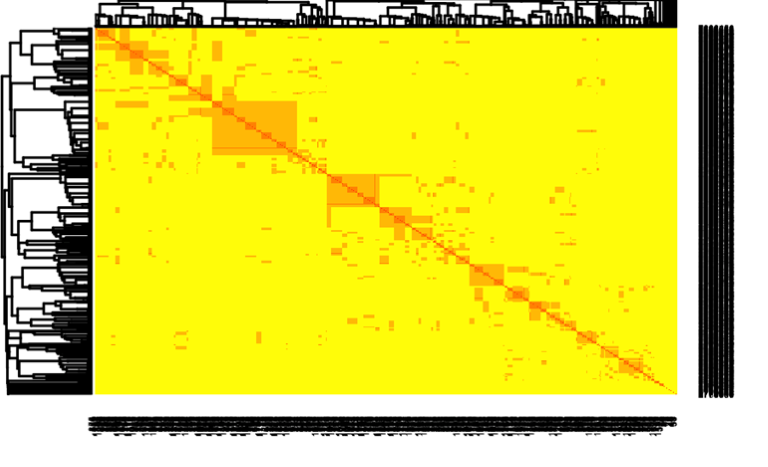
Figur 1: Släktskapsförhållandet baserad på stamtavlan för de individer som ingick i studien. Lodrät axel visar trädindividerna som mödrar och vågrät axel visar trädindividerna som fäder. Mörkare rött visar högre släktskap än ljusröda partier. Den mörka ”linjen” visar släktskapet mellan individerna själva, dvs inavel. De gula partierna visar på obesläktade individer.
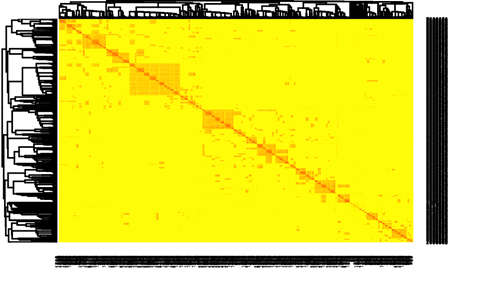
Figur 2: Släktskapsförhållandet baserat på SNP för de individer som ingick i studien. Lodrät axel visar trädindividerna som mödrar och vågrät axel visar trädindividerna som fäder. Mörkare rött visar högre släktskap än ljusröda partier. Den mörka ”linjen” visar släktskapet mellan individerna själva, dvs inavel. De gula partierna visar på obesläktade individer.
När släktskapet baserad på stamtavla (figur 1) jämfördes med släktskapsförhållandet baserad på markördata (figur 2) så hittades inga direkta fel i korsningsarbetet eller det använda sättet att identifiera och hålla isär familjer i plantskolan, det vill säga markörerna stödjer vårt arbetssätt och de stamtavlor som används. Däremot när säkerheten jämfördes mellan de bägge metoderna så visade det sig att man här får säkrare värden när stamtavlan används än om markördata används. Detta trots att utvärderingar där man tar hänsyn till den rekombination som sker vid reproduktiv delning borde vara mer exakt än om man enbart använder sig av släktskapet där man antar att alla syskon är lika besläktade (Ashraf mf.l., 2016).
Möjligheter till faktisk selektion
Resultatet i den här studien stöds av ett flertal andra studier som är gjorda på träd där de använt sig enbart av släktskap baserad på markördata (Beaulieu m.fl, 2014; El-Dien m. fl., 2015; Lenz m. fl., 2017; Resende m. fl., 2017). Inom den svenska skogsträdsförädlingen idag så tar vi enbart hänsyn till den delen av genetiken som ärvs från generation till generation, den additiva variansen, inte vilka föräldrar i kombination som producerar de bästa individerna, icke-additiva variansen. I och med att markördata används så separerars den additiva variansen från den icke-additiva variansen. En orsak till att stamtavlan ger säkrare värden är antagligen att den additiva variansen blir överskattad eftersom modellen inte tar hänsyn till den icke-additiva variansen. Andra studier av den icke-additiva variansens storlek har visat sig ha större betydelse än tidigare antagits (Berlin m.fl.2019). Trots detta finns det ett flertal studier gjorda på träd där markördata givit betydligt högre säkerhet än enbart släktmatris (Müller m. fl., 2017; Tan m. fl., 2017). Vid närmare inspektion av de studierna har felaktigheter hittats i släktskapsmatrisen som antingen kommer från pollenkontaminering eller felmärkning.
Säkerheten blev inte högre med genomiska data, men den blev heller inte nämnvärt lägre trots att enbart genomiska data användes för några individer. Detta gör att det finns en potential att gå vidare för att göra faktisk selektion med genomiska data. I fortsatta studier ska även tas hänsyn till att antalet markörer och antal träd med markördata ständigt ökar vilket gör att noggrannheten kommer att öka. Denna metod kommer även möjliggöra tidigare urval vilket leder till att förädlingen kan gå betydligt snabbare i framtiden. Det är därför mycket intressant att prova metoden över generationer i en faktisk selektion i framtiden. Först då kan metoden inkluderas eller uteslutas i förädlingen.


Vi granskar och publicerar din kommentar så snart som möjligt.
